
Nachhaltigkeit im Klinikbau – In Göppingen entsteht das erste „Green Hospital“ Baden-Württembergs

Die Kliniken Deutschlands durchleben einen massiven Transformationsprozess. Viele Krankenhäuser befinden sich in einer finanziell prekären – teilweise existentiell bedrohlichen – Situation. In dem gesetzlich vorgegebenem Dualen Finanzierungssystem sollen die laufenden Kosten über Fallpauschalen und die notwendigen Investitionen über Fördermittel der Bundesländer gedeckt werden. Seit vielen Jahren kommen die Länder dieser gesetzlichen Vorgabe nicht ausreichend nach, so dass sich bundesweit ein Investitionsstau in der baulichen Struktur vieler Kliniken in Milliardenhöhe angesammelt hat. Die Kliniken sind oftmals gezwungen, notwendige Investitionen und Instandhaltungen aus den laufenden Vergütungen zu finanzieren. Das Defizit der Kliniken Baden-Württembergs summiert sich im forecast auf die Jahresabschlüsse 2024 laut Landkreistagspräsident Joachim Walter auf rund 900 Millionen Euro.
Das ALB FILS KLINIKUM, in hundertprozentig kommunaler Trägerschaft des Landkreises Göppingen, hat sich vor mehr als 10 Jahren gegen die umfassende Sanierung eines Klinikgebäudes zugunsten eines kompletten Neubaus entschieden. Neben essenziellen wirtschaftlichen Gründen standen hier insbesondere prozessuale Optimierungen, die Attraktivität als Arbeitgeber und umfassender Gesundheitsdienstleister sowie die zu erwartenden Neustrukturierungen in der Krankenhauslandschaft im Vordergrund. Das neue Klinikum befindet sich nun im „Endspurt“ der Fertigstellung und wird sich in Teilen über eine „Effizienzrendite“ im Abgleich zum Weiterbetrieb der mehr als 50 Jahre alten Bestandsklinik refinanzieren. Der Grund dafür liegt unter anderem in der strategischen Prämisse des nachhaltigen und hoch energieeffizienten Bauens. Die erwartete Energiepreisentwicklung der kommenden Wirtschaftsperioden wird diese Effizienzrendite des Neubaus weiter steigern.
Raum für modernste prozessoptimierte Medizin und Pflege

Abb. 1: Neubau ALB FILS KLINIKUM, ©Max Radloff Photography
Derzeit entsteht mit dem Neubau des ALB FILS KLINIKUMs in Göppingen eine der modernsten Kliniken in Europa. Mit einem Investitionsvolumen von rund 360 Millionen Euro wird auf 43.000 qm Nutzfläche und sieben Vollgeschossen Raum für modernste prozessoptimierte Medizin und Pflege geschaffen. Mit Baubeginn im Jahr 2019 entstehen dort unter anderem zwölf OP-Säle, inklusive Hybrid OP, drei Herzkatheterlabore, vier Kreißsäle, zwei Intensiv- und eine IntermediateCare-Stationen und ein Hubschrauberlandeplatz. Mit 645 Normalstationsbetten, modernster medizinischer Ausstattung sowie hellen und modernen Räumlichkeiten ist der bauliche Grundstein für eine optimale Behandlungsqualität der Patienten gelegt und auch das Personal profitiert von einer positiven Arbeitsplatzgestaltung.
Von der DGNB mit dem Vorzertifikat in Gold ausgezeichnet
Die Deutsche Gesellschaft für Nachhaltiges Bauen (DGNB) bewertet die Nachhaltigkeit eines Gebäudes und hat den Neubau des ALB FILS KLINIKUMs mit dem DGNB-Vorzertifikat in Gold ausgezeichnet. Die DGNB bestätigt damit, dass eine besonders umweltfreundliche, wirtschaftliche, effiziente, ressourcensparende und optimale Gebäudeplanung für den Neubau verfolgt wurde. Das ALB FILS KLINIKUM ist somit das erste „Green Hospital“ in Baden-Württemberg. Das Klinikum hat, der Unternehmensstrategie folgend, bereits vor Baubeginn und zu einem sehr frühen Planungsstadium diesen Ansatz konsequent verfolgt. Ziel war eine nachhaltige Planung und Umsetzung des Neubaus. Der Antrieb bestand im Besonderen darin, die Wirtschaftlichkeit zu erhöhen und mehr Lebensqualität bei gleichzeitig geringeren Betriebskosten zu erreichen – immer mit dem Fokus auf Mensch und Mitwelt.
Nachhaltigkeitsgedanke im Planungsprozess von Anfang an berücksichtigt
Durch die frühe Einstiegsphase konnten bereits wesentliche Optimierungspotenziale in der Grobplanung des Großprojektes berücksichtigt werden. So konnte beispielsweise eine Reduzierung des Wärmebedarfs um circa 60 Prozent sowie die Umsetzung einer Wärmerückgewinnung mit einem Effizienzgrad von über 85 Prozent realisiert werden. Darüber hinaus wurden die Voraussetzungen für weitere Förderungen durch den Bund geschaffen, indem das Haus am Ende des Planungs- und Optimierungsprozesses den extrem hohen Anforderungen des KfW-55-Programms entsprach.
Die Herausforderungen waren groß, aber machbar

Abb. 2: Neubau ALB FILS KLINIKUM, ©Max Radloff Photography
Die größte Herausforderung bei der Berücksichtigung des nachhaltigen Ansatzes sind die höheren Kosten für nachhaltige Materialien. Beim Neubau des ALB FILS KLINIKUMs wurde insbesondere die Gebäudehülle nach KfW-55-Standard gebaut – dies war zu Baubeginn der höchste Standard unterhalb des Standards des Passivhauses. Verwirklicht wird außerdem eine komplexe Wärmerückgewinnung über Wärmetauscher. Ein gutes Raumklima wird durch eine „Betonkernaktivierung“ der Decken erreicht und durch die Auswahl besonders nachhaltiger Materialien mit positiven Eigenschaften, wie zum Beispiel geringe Ausdünstung, Langlebigkeit, Ursprung aus nachhaltigen Quellen und geringe Schadstoffbelastung. Ein weiterer wichtiger Baustein zur nachhaltigen Bauweise ist die PV-Anlage mit 425 kwh/p und einer hundertprozentigen Nutzung für den Eigenverbrauch. Grundsätzlich umfasst der Green-Hospital-Ansatz nach DGNB ökologisch-ökonomische, soziokulturelle, technische, prozessuale und standortbezogene Qualitäten und Kernbereiche.
Die positiven Effekte sind vielfältig
Durch die nachhaltige Bauweise erhofft sich das Klinikum, seine Attraktivität für Arbeitnehmer weiter zu steigern, denn die neuen Arbeitsplätze wurden mit viel Tageslicht und kurzen Wegen geplant und realisiert. Weiter sieht das Klinikum einen Wettbewerbsvorteil gegenüber anderen Kliniken bei der Patientengewinnung. Günstigere Bankkredite und Tilgungszuschüsse sind ebenso ein wesentlicher Aspekt, wie auch die Vorreiterrolle für andere Unternehmen. Ein positives Image in der Wahrnehmung der Öffentlichkeit ist bereits heute zu spüren.
Zudem stehen für diese nachhaltige Bauweise KFfW-55-Förderungmittel von der Bundesrepublik Deutschland zur Verfügung. Nach der Vorzertifizierung soll nun abschließend das DGNB-Zertifikat in Gold erreicht werden.
Bereits heute ist das ALB FILS KLINIKUM ein KLIMAWIN-Unternehmen des Umweltministeriums Baden-Württemberg und orientiert sich an den Sustainable Development Goals (SDGs) der Agenda 2030 der United Nation (UN). Die DGNB unterstützt ebenso diese Ziele. Um den Zusammenhang einer nachhaltigen Bauweise mit den SDGs herauszuarbeiten und transparent zu machen, sind sämtliche Kriterien zu den Zielen der UN überprüft. Als Ergebnis erhält jedes Projekt, das eine DGNB-Zertifizierung erfolgreich abschließt, künftig eine Aussage darüber, inwieweit es einen Beitrag zur Erreichung der SDG geleistet hat.
Nachhaltiges Bauen zahlt sich langfristig aus
Vor dem Hintergrund der Entwicklung der Nachhaltigkeit in den vergangenen Jahren und der weiteren Fokussierung der Gesellschaft auf dieses Thema war es in den Planungsjahren des Neubaus, 2014 bis 2018, völlig richtig, den Weg des „Green Hospital“ einzuschlagen. Bereits vor Bezug des Neubaus ist klar, dass das Gebäude in Sachen Nachhaltigkeit, Wirtschaftlichkeit und Flexibilität hohe Maßstäbe setzt – auch mit einem konsequenten Fokus auf biologische und nachhaltige Baustoffe. Hohe Wertstabilität sowie Transparenz bei den eingesparten Ressourcen und Emissionen sind gesetzt. Der Ansatz des nachhaltigen Bauens und der Wunsch, der eigenen Verantwortung gegenüber der Umwelt und ökologischen Werten nachzukommen, führten zu einem langen und manchmal schwierigen, Weg. Anderen Kliniken kann an dieser Stelle nur geraten werden, sich nicht entmutigen zu lassen. Bei der Planung von ähnlichen Bauprojekten rät das Klinikum, den CO2-Ausstoß bei den Entscheidungen mit „einzupreisen“ und in der Betrachtung über die reinen Baukosten hinauszugehen, denn das „Green Hospital“ muss in seinem gesamten Lebenszyklus der Immobilie gesehen werden – Langfristigkeit macht sich hier bezahlt.
Mehr Unterstützung würde helfen
Eine Anregung geht an die Politik auf Bundes- und Landesebene: Hier ist mehr Unterstützung wünschenswert. Bei den aktuellen finanziellen Problemen geht es vielen Kliniken rein ums Überleben. Ohne tiefgreifende Förderprogramme können sie Nachhaltigkeit nicht ausreichend berücksichtigen. Die Politik sollte das Engagement in Nachhaltigkeit und Zukunftsfähigkeit öffentlicher Infrastrukturen auch im Bereich „Green Hospital“ stärker fördern, zum Beispiel durch die Bereitstellung von Beratungs- und Zertifizierungspartnern über Bund und Länder. Dies würde auch längerfristige Transformationsprozesse hin zum „Green Hospital“ bei Bestandsimmobilien ermöglichen. So könnte das „Green Hospital“ zum Standard-Hospital werden – zumindest für Neubauten wäre es wünschenswert. Durch die aktuellen Rahmenbedingungen im Gesundheitswesen und den hohen Kostendruck im System wird es sich aber ohne weitere Anreiz- und Fördersysteme nur sehr langsam etablieren können. Der Fokus im Gesundheitssystem muss sich grundsätzlich aber auch an den Vorgaben und Klimazielen der UN und der EU orientieren. Auch die langfristige Unterstützung von öffentlicher Seite ist zwingend notwendig. Der öffentliche Gesundheitssektor wird diesen tiefgreifenden Transformationsprozess nur mit Unterstützung durch Bund und Länder leisten können. Ein gutes Beispiel könnten hier Vergleichsprojekte, wie beispielsweise die Digitalisierung mit der Förderung durch das KrankenhausZukunftsGesetz (KHZG) sein.
Über die Autoren:
Dr. Ingo Hüttner ist medizinischer Geschäftsführer sowie Vorsitzender der Geschäftsführung der ALB FILS KLINIKUM GmbH. Seit 2021 gehört Dr. Hüttner dem Vorstand der DGQ an.
Wolfgang Schmid ist Kaufmännischer Geschäftsführer der ALB FILS KLINIKUM GmbH.
Bildnachweis: ©Max Radloff Photography
Grüne Gesundheit: Nachhaltigkeit und Klimaschutz im Gesundheitswesen

In den letzten Jahren haben die Themen Nachhaltigkeit und Klimaschutz in nahezu allen Lebensbereichen an Bedeutung gewonnen. Besonders im Gesundheitswesen, einem der größten und ressourcenintensivsten Sektoren, ist die Notwendigkeit für Maßnahmen in Richtung nachhaltigeren Handelns evident. Der Klimawandel hat nicht nur für den Planeten drastische Folgen, sondern auch für die menschliche Gesundheit. Die klimatischen Veränderungen begünstigen die Zunahme von Allergien, Erkrankungen der Atemwege und des Herz-Kreislauf-Systems, sie führen zu steigenden Infektionszahlen und zur Zunahme von psychischen Erkrankungen wie Depressionen. Nachhaltigkeit und Klimaschutz anhaltend in den Fokus des Handelns im Gesundheitswesen zu rücken, ist somit von großer Relevanz.
Doch was bedeutet „Nachhaltigkeit“ und welche Ansätze gibt es, um Gesundheitseinrichtungen wie Krankenhäuser nachhaltiger und das Handeln im Einklang mit den Bemühungen um Klimaneutralität zu gestalten?
Die drei Dimensionen der Nachhaltigkeit
Das Bundesministerium für wirtschaftliche Zusammenarbeit und Entwicklung versteht den Begriff als ethisches Handlungsprinzip, das uns verpflichtet, „die Bedürfnisse der Gegenwart so zu befriedigen, dass die Möglichkeiten zukünftiger Generationen nicht eingeschränkt werden.“ Konkretisiert wird diese recht vage Definition durch das Drei-Säulen-Modell. Die drei Säulen bilden
- die Ökologie,
- die Ökonomie und
- das Soziale.
Der Parlamentarische Beirat für nachhaltige Entwicklung des Bundestages formuliert eine konkretere Handlungsmaxime: „Wir dürfen nicht heute auf Kosten von morgen leben! Wir sollen nicht mehr verbrauchen, als künftig wieder bereitgestellt werden kann.“ Angesichts der massiven Effekte des Klimawandels und den Auswirkungen für zukünftige Generationen liegt der Fokus der Diskussionen um Nachhaltigkeit und nachhaltiges Handeln auf der Säule „Ökologie“.
Nachhaltigkeit und Klimaschutz im Gesundheitswesen: Status Quo
Der Gesundheitssektor in Deutschland ist ein Wirtschaftszweig mit großer ökonomischer Bedeutung und spielt eine zentrale Rolle bei der Gesundheitsversorgung der Bevölkerung. Im Jahr 2022 betrugen die Gesundheitsausgaben in Deutschland 497,7 Milliarden Euro, was etwa 12,8 Prozent des Bruttoinlandsprodukts (BIP) ausmachte. Derzeit arbeiten rund 6 Millionen im Gesundheitswesen. Das entspricht etwa 17,7 Prozent der Gesamtbeschäftigung. Jeder achte Erwerbstätige in Deutschland ist somit in einer Einrichtung des Gesundheitswesens beschäftigt. Die Tendenz ist schon seit Jahren steigend.
Krankenhäuser und andere Gesundheitseinrichtungen verbrauchen global große Mengen an Energie und Wasser, erzeugen erhebliche Mengen an Abfall und emittieren eine Vielzahl von Schadstoffen. Die Bundesärztekammer berichtet mit Verweis auf den „Health care climate footprint report“, dass der Gesundheitssektor mit zwei Gigatonnen CO2 Äquivalent pro Jahr für 4,4 Prozent der globalen Nettoemissionen verantwortlich ist. Zur besseren Einordnung dieser Zahl bietet die Kammer einen anschaulichen Vergleich: „Wäre der globale Gesundheitssektor ein Land, wäre er der fünftgrößte Emittent von Klimagasen im weltweiten Ranking der Länder.“ Das Gesundheitswesen verursacht aktuell gar mehr Schadstoffemissionen als der Flugverkehr oder die Schifffahrt, zu dem Ergebnis kommt das von PwC veröffentlichte Healthcare-Barometer 2022. Mit rund 71 Prozent verursachen Medizinprodukte und die mit ihnen verbundenen Lieferketten den größten Anteil der Emissionen. Mit rund 5 Prozent des Gesamtrohstoffkonsums in Deutschland liegt das Gesundheitswesen auch in diesem Bereich weit vorne.
Die Aktivitäten des Gesundheitswesens tragen somit maßgeblich zur Belastung unseres Klimas und der Umwelt bei. Zugleich bedeutet dies, dass durch das Ergreifen geeigneter Maßnahmen ein signifikanter Beitrag für mehr Nachhaltigkeit und Klimaschutz beleistet werden kann.
Strategien und Maßnahmen zur Förderung der Nachhaltigkeit und des Klimaschutzes
Welche Maßnahmen sind nun geeignet, um Nachhaltigkeit und Klimaschutz im Gesundheitswesen anhaltend zu verankern? Es gibt eine Vielzahl von teilweise niedrigschwelligen Möglichkeiten und verschiedenen Ansatzpunkte. Um den Unternehmen den ersten Schritt in Richtung Handeln zu erleichtern, hat die Arbeitsgruppe „Klimawandel“ der Bundesärztekammer zehn Handlungsfelder identifiziert und für jedes Empfehlungen erarbeitet.
Die folgende Auflistung soll einen Eindruck vermitteln und einen Überblick geben:
- Unternehmensführung, bspw. Etablierung eines Berichtswesens zum CO2-Fußabdruck und zu den ergriffenen Maßnahmen, um den CO2-Ausstoß zu reduzieren.
- Energieverbrauch, bspw. Optimierung des Einsatzes von Energie zur Warmwassererzeugung durch Einsatz erneuerbarer Energien und durch Einsatz wassersparender Steuerungsmechanismen
- Gebäude und Gelände, bspw. Ausweitung von Grünanlagen inklusive Förderung der Biodiversität, klimaadaptierte Baumpflanzung
- Anästhesiegase / Inhaler / chemische Stoffe, bspw. Auffangen des freiwerdenden CO2 durch Filter, Reduktion von Inhalationssprays / Inhaler wo möglich durch Nutzung anderer Darreichungsformen der Medikamente
- Wasser, bspw. Minimierung des Wasserverbrauchs durch Sammeln von Regenwasser zur Bewässerung von Gartenanlagen
- Abfall, bspw. Reduktion der Verwendung von Einmalprodukten, Umstellung auf Recyclingpapier, Papiervermeidung durch Digitalisierung
- Transport, Vermeidung unnötiger Fahrten/Reisen durch Videokonferenzen und -sprechstunden, Teilnahme an Programmen zum Fahrradleasing
- Einkauf, bspw. Optimierung der Lieferketten
- Ernährung, bspw. vermehrte Nutzung lokaler und saisonaler Produkte für Patienten, Mitarbeiter und Besucher, vermehrt vegetarische Küche
- Büro / EDV, bspw. Nachhaltigkeit im Internet bei der Auswahl von Suchmaschinen und E-Mail-Diensten
Selbstredend dürfen die Maßnahmen die Einhaltung medizinischer Standards nicht gefährden. Das Patientenwohl ist nach wie vor das oberste Gut im Gesundheitswesen. In vielen Fällen sind die Maßnahmen zunächst mit Investitionen verbunden. Mittel- und langfristig können die Unternehmen jedoch anhaltend ihre Kosten reduzieren und möglicherweise ihre Prozesse effizienter gestalten.
Die Treiber für mehr Nachhaltigkeit und Klimaschutz im Gesundheitswesen
Die Einführung nachhaltiger Praktiken im Gesundheitswesen wird von einer Vielzahl von Faktoren und Treibern beeinflusst. Sie lassen sich in verschiedene Kategorien einteilen, die alle dazu beitragen, dass der Sektor ökologisch verantwortungsvoller wird.
- Medizinischer und technologischer Fortschritt:
Moderne Technologien wie energieeffiziente medizinische Geräte, digitale Gesundheitslösungen und telemedizinische Anwendungen haben nicht nur das Potential, die Qualität von und den Zugang zu Gesundheitsdienstleistungen zu steigern, sondern zugleich den Ressourcenverbrauch drastisch zu senken. Beispielsweise ermöglichen digitale Patientendaten eine effizientere Verwaltung bei verringertem Papierverbrauch.
- Demografische Veränderungen und steigende Gesundheitskosten:
Die alternde Bevölkerung in vielen Industrieländern führt zu einer erhöhten Nachfrage nach Gesundheitsdienstleistungen. Diese steigende Nachfrage übt Druck auf die Gesundheitssysteme aus und macht effiziente, nachhaltige und kostensensitivere Lösungen notwendig. Nachhaltige Praktiken, die beispielsweise den Energie- und Ressourcenverbrauch reduzieren und Abfall minimieren, tragen wesentlich dazu bei, die Betriebskosten der Einrichtungen zu senken.
- Gesetzliche und regulatorische Anforderungen:
Regierungen und internationale Organisationen arbeiten stetig an der Umsetzung neuer Vorschriften und Standards und stellen zunehmend strengere Anforderungen an die Einrichtungen des Gesundheitswesens. Die Richtlinie (EU) 2022/2464 zur Nachhaltigkeitsberichterstattung beispielsweise, die am 5. Januar 2023 in Kraft getreten und bis Mitte dieses Jahres in nationales Recht umzusetzen ist, bedeutet für die Unternehmen umfassende Änderungen der Anforderungen hinsichtlich der nichtfinanziellen Berichterstattung.
Die Hürden für mehr Nachhaltigkeit und Klimaschutz im Gesundheitswesen
Während der demografische Wandel, die zunehmende Digitalisierung und der technologische Fortschritt einerseits Treiber für mehr Nachhaltigkeit und Klimaschutz sein können, stellen sie Gesundheitseinrichtungen zugleich vor große Herausforderungen. Viele Häuser schaffen es angesichts der vielfältigen Anforderungen nicht, das in den Trends liegende Potenzial für ihre Organisation zu heben. Zugleich hemmen Entwicklungen wie der Fachkräftemangel die Verankerung von Nachhaltigkeit und Klimaschutz: Ein vom Bundesgesundheitsministerium in Auftrag gegebenes Gutachten nennt zudem die folgenden Faktoren als wesentliche Hemmnisse:
- Fehlende Anreize und Informationen: Angesichts fehlender monetärer oder regulatorischer Anreize entscheiden sich Unternehmen oft, sich nicht mit der Umsetzung von Maßnahmen zu beschäftigen. Zugleich fehlen in vielen Fällen Informationen zur Finanzierung von Nachhaltigkeitsmaßnahmen.
- Organisatorische Einschränkungen: Belange der Nachhaltigkeit werden in der Regel nicht im Management thematisiert und sind nur wenig in den Leitungsebenen der Unternehmen verankert.
- Ressourcenmangel: Es mangelt den Einrichtungen an Zeit, Geld und Personal.
Fazit
Wenngleich es praxistaugliche und teils niedrigschwellige Ansatzpunkte gibt, wird im Gesundheitswesen aktuell zu wenig Gewicht auf nachhaltige Praktiken gelegt. Um Nachhaltigkeit dauerhaft in den Prozessen zu verankern, bedarf es weiterer Impulse und regulatorischer Vorgaben, sowie der Platzierung des Themas auf Leitungsebene. Die Effekte des Klimawandels werden zunehmend deutlich. Die Bedrohungen für Gesellschaft und Umwelt betreffen alle. Jeder Akteur steht in der Pflicht, einen Beitrag zu leisten. Diese Sicht teilt auch die Ärzteschaft. Der 125. Deutsche Ärztetag konstatierte die Notwendigkeit einer nationalen Strategie für eine klimafreundliche Gesundheitsversorgung und fordert Klimaneutralität bis 2030. An geeigneten Maßnahmen fehlt es nicht. Seitens der Politik bedarf es jedoch der Schaffung der notwendigen Rahmenbedingungen und seitens der Unternehmen des Umsetzungswillens. So oder so, die Effekte des Klimawandels werden sich auch in Zukunft zunehmend verschärfen und die Menschen begleiten.
Über die Autorin:
Nathalie Roskaritz ist Produktmanagerin und in der DGQ Weiterbildung für die Angebote im Bereich Qualitätsmanagement im Gesundheits- und Sozialwesen verantwortlich.
Nachvollziehbare und verlässliche Ergebnisse durch Computer System Validierung
Frederik Janas ist DGQ-Trainer des neuen E-Trainings „Computer System Validierung (CSV)“ für die Medizinprodukteindustrie und Experte für Medizinproduktesicherheit bei CRConsultants. Im Interview erläutert er, warum das Thema „Computer System Validierung“ im Kontext der Herstellung von sicheren Medizinprodukten so wichtig ist.
Herr Janas, der Begriff „Computer System Validierung“ ist sicher nicht jedem geläufig. Könnten Sie uns erklären, was er bedeutet?
Frederik Janas: Computer System Validierung bezeichnet die Prüfung und den dokumentierten Nachweis, dass ein Computersystem oder eine eigenständige Software spezifische Anforderungen erfüllt. Initial werden Spezifikationen definiert, die sowohl allgemeine Anforderungen als auch spezifische Benutzerfälle darstellen. Im weiteren Verlauf der CSV werden Nachweise in Form von Tests geplant, durchgeführt und dokumentiert. Anders ausgedrückt, vorab wird definiert, wie ein Computersystem oder eine Software funktionieren soll und im Rahmen der Validierung wird geprüft, ob diese Funktion verlässlich und korrekt erfüllt wird.
Welche Arten von Software oder Computersystemen gibt es im Kontext der Medizinprodukteherstellung und müssen sie alle validiert werden?
Frederik Janas: Es können zwei verschiedene Arten von Computer Systemen beziehungsweise Software unterschieden werden. Erstens solche, die in der Produktion von Medizinprodukten oder Dienstleistungserbringung und für das Qualitätsmanagementsystem zum Einsatz kommen, Datenanalysesoftware zum Beispiel. Diese sind gemäß den Anforderungen der ISO 13485 zu validieren.
Zweitens solche, die selbst eigenständige Medizinprodukte – Software as a Medical Device (kurz SaMD) – oder Teil eines Medizinprodukts sind. Zum Beispiel könnte das Software sein, die für Überwachungs- oder Diagnosesysteme benötigt wird. Diese Software muss gemäß der ISO 62304 beziehungsweise ISO 82304 validiert werden.
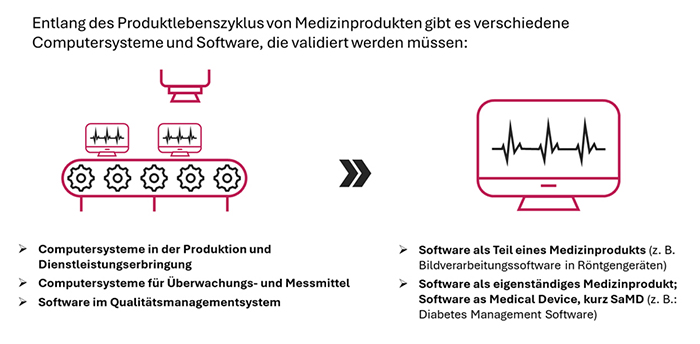
Abb. 1: Software- und Computersysteme im Kontext der Medizinprodukteherstellung
Warum ist die Validierung von Computersystemen so wichtig?
Frederik Janas: Zum einen müssen regulatorische Anforderungen eingehalten werden, der risikobasierte Ansatz der CSV ist beispielsweise normativ durch die ISO 13485 gefordert, die unter anderem bei Audits durch Benannte Stellen überprüft werden. Wir konnten feststellen, dass die CSV in den letzten Jahren verstärkt in den Fokus der Auditor:innen gerückt ist. Außerdem trägt die CSV zur Sicherheit und Einhaltung der Qualität von Medizinprodukten im Zusammenhang mit der Anwendung von Softwaresystemen bei. Da die Funktionen von heutigen Softwaresystemen immer komplexer werden, wird es immer schwieriger, nachvollziehbare Ergebnisse zu erzielen. Eine CSV hilft durch strukturierte Verfahren, die Validität der Ergebnisse zu beweisen.
Könnten Sie uns einen Überblick darüber geben, welche verschiedenen Arten der Validierung es gibt?
Frederik Janas: Neben der Computer System- beziehungsweise Softwarevalidierung werden Validierungen auch für Testmethoden, Umgebungsbedingungen sowie Prozesse und Ausrüstungen durchgeführt. Auch hier geht es immer darum sicherzustellen, dass die definierten Anforderungen verlässlich erfüllt werden. Beispielsweise zielt die Prozessvalidierung darauf ab, die Wirksamkeit der Prozesse, die gegebenenfalls im Zusammenhang mit der Softwareentwicklung- und -anwendung stehen, zu überprüfen. Im Rahmen der Digitalisierung ist Software ein häufiger Bestandteil in der Produktion oder bei der Überwachung von Anforderungen. Somit kann es bei allen Arten der Validierungen Schnittstellen zur Computer System Validierung geben.
Welche Konsequenzen können auftreten, wenn eine angemessene Computer System Validierung nicht durchgeführt wird?
Frederik Janas: Regulatorisch kann dies Konsequenzen in Form von Abweichungen bei Audits nach sich ziehen. Zudem kann durch fehlerhafte Funktionen die Produktqualität und Patientensicherheit beeinträchtigt werden. Mögliche Auswirkungen aufgrund nicht valider Computersysteme können falsche Parameter in der Produktion, falsche Angaben auf Produkten, falsche Ergebnisse in der Überwachung von Medizinprodukten oder mangelnde Rückverfolgbarkeit im Qualitätsmanagementsystem sein.
Lassen Sie uns die Praxis gemeinsam beleuchten. Welche Schritte umfasst der Validierungsprozess konkret?
Frederik Janas: Allgemein sind die Schritte der Validierung gemäß V-Modell in das Definieren von Anforderungen, das Testen von Funktionen und das Dokumentieren der Kriterien und Ergebnisse unterteilt. Der Umfang ist stark abhängig von der Komplexität der Anwendung sowie dem Risiko. Hier liefert die Software-Kategorisierung gemäß GAMP 5 (Good Automated Manufacturing Practice) eine gute Richtlinie, in der Software abhängig von der Standardisierung und Konfiguration eingestuft wird. Das bedeutet, dass in der Praxis die Schritte individuell definiert werden. Für eine einfache Software wie beispielsweise ein Excel-Sheet umfasst der Validierungsprozess gegebenenfalls nur einen Prüfplan, in dem die Beschreibung, Anforderung, Methodik und das Risiko beschrieben sind, sowie einen Prüfbericht, der die Ergebnisse darstellt. Für eine komplexe Software mit hohem Risiko sind mehr Schritte notwendig. Hierbei kann initial eine Risikobewertung durchgeführt und die Anforderungen in verschiedenen Detaillierungsstufen definiert werden. Daraus resultieren häufig mehrere Prüfpläne und -berichte.
Und zu guter Letzt, welche Werkzeuge und Methoden werden für die Validierung von Computersystemen eingesetzt?
Frederik Janas: Das wichtigste Werkzeug ist eine Softwaremasterliste sowohl aus interner Sicht als auch aus Sicht eines externen Auditors. Diese schafft eine Übersicht aller verwendeten Computersysteme inklusive weiterer Angaben unter anderem zur Validierungspflicht. Weiterhin ist die Risikobewertung in Form einer initialen Bewertung oder FMEA ein wichtiges Werkzeug, um die Validierungstätigkeiten risikobasiert zu priorisieren. Während der Validierung werden verschiedene Testmethoden herangezogen. So zum Beispiel Positiv-, Wiederholungs- und Regressionstests. Diese werden spezifisch für den Test ausgewählt und sollen einen geeigneten Nachweis zur Erfüllung der Anforderungen erbringen.
Die deutsche Medizinprodukte-Branche: vom Mittelstand geprägt, von Innovation getragen
Die Medizinprodukte-Branche nimmt als Akteur der industriellen Gesundheitswirtschaft eine wichtige Rolle ein. Sie ist zudem beispielhaft für die hohe Bedeutung des Mittelstands in der deutschen Wirtschaft. Neben dem Beitrag, den sie für die Sicherstellung und Weiterentwicklung qualitativ hochwertiger Patientenversorgung leistet, bietet sie großes wirtschaftliches Potenzial. Doch Lieferschwierigkeiten, politische Unwägbarkeiten und andere Herausforderungen ziehen auch an der mittelständisch geprägten Medizinprodukte-Branche nicht folgenlos vorbei. Auch branchenspezifische Entwicklungen, wie die Änderungen in der EU-Medizinprodukte-Verordnung (MDR), setzen insbesondere die kleinen und mittelständischen Unternehmen weiterhin unter Druck.
International als verlässlicher Zulieferer bekannt
Technologische Durchbrüche, die Zunahme von Zivilisationskrankheiten wie Diabetes, der demografische Wandel und der mit diesen Entwicklungen einhergehende stetig wachsende klinische Versorgungsbedarf – die Faktoren die dazu führen, dass Medizinprodukte zunehmend an Bedeutung gewinnen, sind vielfältig. Die große Bandbreite der Medizinprodukte umfasst Verfahren und Lösungen für Diagnostik und Therapie. Sie leisten in vielerlei Hinsicht einen wichtigen Beitrag: Zum einen tragen sie maßgeblich zur Leistungsfähigkeit des deutschen Gesundheitssystems und zur Patient:innenversorgung bei. Ob Lesebrille, Katheter, Implantate oder Röntgengerät – für Patient:innen bedeuten die verschiedenen Produkte konkret gesteigerte Lebensqualität, in vielen Fällen sogar lebensrettende Therapie. Zum anderen ist die die deutsche Medizinprodukte-Branche mit über 400.000 verschiedenen Produkten und einem Gesamtumsatz von 38,4 Milliarden Euro im Jahr 2022 ein wachstumsstarker und heterogener Wirtschaftszweig. Angesichts der über 250.000 Beschäftigten ist sie zudem aus beschäftigungspolitischer Perspektive von großem Interesse. Doch nicht nur national, auch international erfreuen sich deutsche Medizinprodukte großer Beliebtheit. Mit einer Exportquote von 67 Prozent im Jahr 2022 sind die deutschen Unternehmen auf dem internationalen Markt als verlässliche Zulieferer von qualitativ hochwertigen Produkten bekannt. Die im internationalen Vergleich starke Stellung zeugt davon, dass Deutschland als guter Standort für Innovation und Produktion geschätzt wird.
Hohe Relevanz kleiner und mittlerer Unternehmen
Die Branche ist stark mittelständisch geprägt. Die Betrachtung der Betriebsverteilung nach Beschäftigtengrößenklassen zeigt, dass rund 93 Prozent der Unternehmen, rund 1.300 Betriebe, weniger als 250 Mitarbeiter:innen beschäftigen und somit zur Kategorie der kleinen und mittleren Unternehmen (KMU) zählen. Der Anteil der Unternehmen mit mehr als 250 Mitarbeiter:innen beläuft sich indes auf nur 7 Prozent. 4 Prozent davon sind Unternehmen mit 250 bis 499 Mitarbeiter:innen, 3 Prozent mit 500 und mehr.
Doch ist der Mittelstand nicht nur aufgrund seiner Betriebsstärke von entscheidender Bedeutung für die Medizinprodukte-Branche:
- Agilität und Flexibilität: KMU sind in ihrem Handeln oftmals agiler und flexibler als große Konzerne. Sie können schneller auf neue Marktanforderungen und technologische Entwicklungen reagieren und tragen so zu einem dynamischen Markt bei.
- Nähe zum Markt: KMU sind oft in engerem Kontakt mit dem Markt, als größere Unternehmen und sind daher besser vertraut mit den Schmerzpunkten und Bedürfnissen ihrer Kunden. So können sie effektiver auf spezifische Anforderungen reagieren und Produkte entwickeln, die präzise auf diese Bedürfnisse zugeschnitten sind.
- Spezialisierung: Viele KMU in der Medizinprodukte-Branche haben ein feines Gespür für Nischenmärkte. Dies ermöglicht es ihnen, hochspezialisierte Lösungen zu entwickeln, die von größeren Unternehmen möglicherweise nicht verfolgt werden.
KMU sind insofern maßgeblich für die Vielfalt verantwortlich, für die die deutsche Medizinprodukte-Branche bekannt ist. Sie sind zudem von einer starken Innovationskultur geprägt und fördern Kreativität und Austausch.
Anspruchsvolles Innovationsumfeld
Die deutsche Medizinprodukte-Branche ist hoch innovativ und von kurzen Produktzyklen geprägt: Rund ein Drittel des Umsatzes wird in der Medizinprodukte-Branche mit Produkten erzielt, die weniger als drei Jahre alt sind. Im Durchschnitt investieren die Unternehmen rund 9 Prozent ihres Umsatzes in Forschung und Entwicklung. Und das, obwohl Innovation in der Medizinprodukte-Branche deutlich anspruchsvoller ist, als in manch anderer Branche:
- Strenge Regulierung: Die Branche unterliegt strengen regulatorischen und normativen Anforderungen. Für die Entwicklung und Markteinführung neuer Produkte müssen zahlreiche Gesetze und Normen berücksichtigt werden, die beispielsweise hohe Ansprüche an die klinische Erprobung und Dokumentation definieren und so die Sicherheit der Produkte sicherstellen.
- Langwierige Entwicklungszyklen und hohe Kosten: Die Entwicklung neuer Medizinprodukte kann viele Jahre in Anspruch nehmen und ist ausgesprochen kostenintensiv. Dies liegt unter anderem an den umfangreichen formalen Anforderungen an die Entwicklung und Voraussetzungen für die Zertifizierung.
- Komplexe Technologie: Medizinprodukte nutzen oft hochkomplexe Technologien, die spezialisiertes Fachwissen erfordern. Die Integration von Elektronik, Software und Mechanik setzt ein tiefes Verständnis verschiedener Fachgebiete voraus.
Alles in allem erfordert die Innovation in der Medizinproduktebranche ein hohes Maß an Fachwissen, Ressourcen und Zeit.
Chancen und Herausforderungen
Die Branche sieht sich aktuell vielfältigen Herausforderungen gegenüber. Zum einen wirken Trends und Entwicklungen wie die digitale Transformation, Fachkräftemangel und Lieferkettenunsicherheiten sowie die in Folge der Corona-Pandemie gestiegenen Kosten belastend auf die Unternehmen. Zum anderen bedeutet die Änderung in der EU-Medizinprodukte-Verordnung (MDR), etwa die verschärften Anforderungen an die klinische Bewertung, einen wesentlichen Mehraufwand. Diesen können insbesondere die KMU, aber auch die mit der Zulassung betrauten „Benannten Stellen“ in vielen Fällen nicht leisten. Lassen sich hierfür keine pragmatischen Antworten finden, ist zu befürchten, dass die Hersteller ihr Produktportfolio reduzieren. In der Folge wären dringend benötigte Medizinprodukte nicht mehr verfügbar.
Fazit
Deutschland braucht einen zukunftsfähigen Markt und einen starken Mittelstand. KMU sind in der Medizinproduktebranche von zentraler Bedeutung, da sie nicht nur Innovationen vorantreiben, sondern auch zur Vielfalt und vor allem zur Lösung gesundheitlicher Herausforderungen beitragen. Ihre Fähigkeit, schnell zu handeln und auf lokale und globale Bedürfnisse zu reagieren, macht sie zu einem unverzichtbaren Teil der deutschen Wirtschaft.
Über den Autor:
Nathalie Roskaritz ist als Produktmanagerin in der DGQ Weiterbildung für die Angebote im Bereich Medizinprodukte verantwortlich.
Medizinprodukte-Talk im Regionalkreis Berlin
Zum 1. Medizinprodukte-Talk im DGQ-Regionalkreis Berlin am 04.06.2019 kamen trotz hochsommerlicher Temperaturen über 20 Interessierte und DGQ-Mitglieder in die Geschäftsstelle Berlin.

Dieser Termin soll der Auftakt zu einer neuen Veranstaltungsreihe sein, auf der sich Gleichgesinnte mit den Problemen und aktuellen Herausforderungen des Qualitätsmanagements im Bereich der Medizinprodukte befassen möchten.
Die außerordentliche Resonanz und die gut besuchte Veranstaltung zeigten, dass es auch und gerade wegen der neuen europäischen Regulierungen 2017/745/EU (Medical Device Regulation; MDR), die ab 26.05.2020 verbindlich anzuwenden sind, viele Fragen und Bauchschmerzen nicht nur bei Handelsakteuren, also Herstellern, Importeuren und Vertreibern von Medizinprodukten sondern auch bei den unabhängigen Prüfinstitutionen, den Benannten Stellen gibt.
Um hier eine Basis für den Gedankenaustausch zu schaffen, bemühten sich Regionalkreisleiter Peter Stresemann zusammen mit Dr. Claudia Dannehl, Christian Sobotta und Jörg Brokmann in umfangreichen Vorbereitungen, ein Format zu entwickeln, welches hierfür geeignet ist.
Viele Themen wurden gesammelt und andiskutiert, so dass bestimmt bald eine zweite Talkrunde geplant werden kann.
Den ausführlichen Bericht sowie die Präsentation und das Fotoprotokoll finden Sie zum Download auf der DGQ-Mitgliederplattform DGQaktiv.
Neue Medizinprodukte-Verordnung birgt Herausforderungen und Chancen
Der Schrecken der Medizinprodukteindustrie, die neue Medizinprodukte-Verordnung (Medical Device Regulation – MDR), geistert weiterhin durch die Branche. Auch wenn sich der undurchsichtige Schleier an Fragen in manchen Punkten immer mehr lüftet, stehen viele Medizinproduktehersteller vor großen Herausforderungen.
Die neue MDR ist am 25. Mai 2017 in Kraft getreten und soll das bisherige Medizinprodukterecht und die Richtlinien 93/42 und 90/385 ersetzen. Ziel der Änderungen ist es, die Patientensicherheit und die Transparenz der Branche zu steigern.
Jedes Medizinprodukt erhält künftig eine Identifizierungsnummer
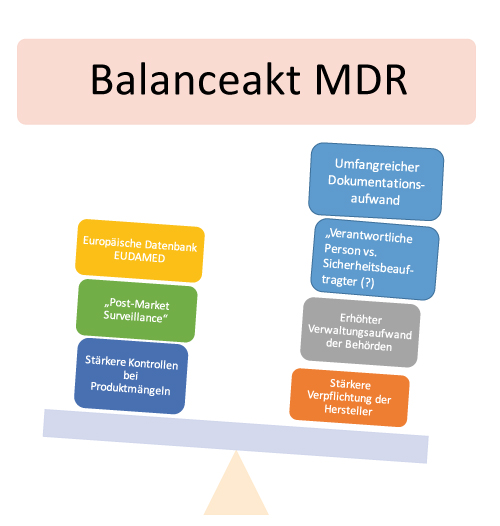 Zukünftig soll eine Expertengruppe in den Zertifizierungsprozess involviert werden, wenn Mängel bei Hochrisiko-Medizinprodukten vorliegen. Außerdem müssen im Rahmen der „Post-Market-Surveillance“ klinische Daten auch nach der Markteinführung gesammelt, dokumentiert und ausgewertet werden. Jedes Medizinprodukt erhält künftig eine Produktidentifizierungsnummer (UDI). Hinzukommt: Eine einheitliche Informationssammlung über umlaufende Medizinprodukte soll die neue Datenbank Eudamed garantieren. Die neue MDR ist eine Herausforderung.
Zukünftig soll eine Expertengruppe in den Zertifizierungsprozess involviert werden, wenn Mängel bei Hochrisiko-Medizinprodukten vorliegen. Außerdem müssen im Rahmen der „Post-Market-Surveillance“ klinische Daten auch nach der Markteinführung gesammelt, dokumentiert und ausgewertet werden. Jedes Medizinprodukt erhält künftig eine Produktidentifizierungsnummer (UDI). Hinzukommt: Eine einheitliche Informationssammlung über umlaufende Medizinprodukte soll die neue Datenbank Eudamed garantieren. Die neue MDR ist eine Herausforderung.
Wirtschaftsakteure stärker in der Pflicht
So haben beispielsweise die Behörden bisher wenig Erfahrung mit der neu geregelten Marktüberwachung. Sie müssen nun stichprobenartig Kontrollen von Produkten und Unterlagen durchführen und die betroffenen Wirtschaftsakteure in die Pflicht nehmen. Die Behörden sind dann dafür zuständig, die Beseitigung der Mängel konsequent zu kontrollieren. Darüber hinaus müssen sie alle beteiligten Stellen informieren und auch Einwendeverfahren verwalten. Bis auf den höheren Verwaltungsaufwand ändert sich aber nicht viel, da das geltende Recht bereits viele Punkte abdeckt. Die Wirtschaftsakteure werden jedoch stärker in die Pflicht genommen, was wiederum neue Ansatzpunkte zur Marktüberwachung durch Behörden ermöglicht.
Rolle der „verantwortlichen Person“ gewinnt an Bedeutung
Kleinere Tücken betreffen beispielsweise die Zukunft des Sicherheitsbeauftragten. Denn die MDR sieht eine „verantwortliche Person“ (responsible person) zur Einhaltung der Regulierungsvorschriften vor. Der Verantwortungsbereich der verantwortlichen Person ist umfassender als der des Sicherheitsbeauftragten. Außerdem ist die Qualifikation spezifischer definiert. Während der Sicherheitsbeauftragte eher über Sachkenntnis verfügen muss, zeichnet die verantwortliche Person eine Fachkenntnis aus. Die Rolle der verantwortlichen Person ist auch deutlich hervorgehobener als die des Sicherheitsbeauftragten, da sie mehr Entscheidungsbefugnisse hat. Je nach Unternehmensgröße dürfen Unternehmen die verantwortliche Person extern benennen. Unklar ist, was das für den bisherigen Sicherheitsbeauftragten bedeutet. Löst die verantwortliche Person den Sicherheitsbeauftragten in Zukunft ab?
Dokumentationsaufwand steigt
Hersteller kritisieren vor allem auch den erhöhten Dokumentationsaufwand von u.a. Entwicklungsakten, Produktakten, Risikomanagementakten. So ist beispielsweise eine umfangreichere klinische Bewertung und ein Nachweis der grundlegenden Anforderungen und Produktbeobachtung notwendig. Die Dokumentation muss immer wieder auf Aktualität geprüft und ergänzt werden. Das frisst Ressourcen.
In den Unternehmen schwirren weiterhin einige Fragezeichen umher. Wer darf eigentlich was? Wer sind die benannten Stellen? Wie wird die MDR von diesen Stellen interpretiert?
Problematisch ist die geringe Anzahl der bisher benannten Stellen. Die Befürchtung ist, dass es nach der Neubenennung der Stellen Anfang 2019 zu einem „Flaschenhals-Effekt“ kommt, da es sowieso seit Jahren eine rückgängige Zahl an benannten Stellen gibt.
Ab dem 26. Mai 2020 ist die MDR verpflichtend anzuwenden. Viele Hersteller kritisieren die nur dreijährige Übergangszeit. Es ist fragwürdig, ob die umfangreichen Anforderungen der MDR in dieser Zeit zu erfüllen sind. Vor allem kleine und mittelständische Unternehmen verfügen oft nicht über die nötigen finanziellen und personellen Ressourcen, um die neuen Anforderungen strukturell im Unternehmen zu verankern.
Gut umgesetzt ist die MDR eine Chance
In den Medien ist immer wieder vereinzelt von Medizinprodukteskandalen zu lesen. Qualitätsmängel an Medizinprodukten sind ein Problem in der Branche und erregen in der Regel viel Aufsehen. Der Grundgedanke der MDR, die Patientensicherheit zu erhöhen und ein europäisches System auf den Weg zu bringen, ist auf jeden Fall positiv. Denn eine Vereinheitlichung und der Versuch, Mängel besser identifizieren zu können, kann unangenehmen Skandalen vorbeugen. Wird die MDR gut umgesetzt, ist sie auf jeden Fall eine Chance, die die Sicherheit der Medizinprodukten fördert. Wie gut diese gute Umsetzung in der knappen Zeit zu bewältigen ist, wird sich erst in naher Zukunft zeigen.

