Neue Medizinprodukte-Verordnung birgt Herausforderungen und Chancen
Der Schrecken der Medizinprodukteindustrie, die neue Medizinprodukte-Verordnung (Medical Device Regulation – MDR), geistert weiterhin durch die Branche. Auch wenn sich der undurchsichtige Schleier an Fragen in manchen Punkten immer mehr lüftet, stehen viele Medizinproduktehersteller vor großen Herausforderungen.
Die neue MDR ist am 25. Mai 2017 in Kraft getreten und soll das bisherige Medizinprodukterecht und die Richtlinien 93/42 und 90/385 ersetzen. Ziel der Änderungen ist es, die Patientensicherheit und die Transparenz der Branche zu steigern.
Jedes Medizinprodukt erhält künftig eine Identifizierungsnummer
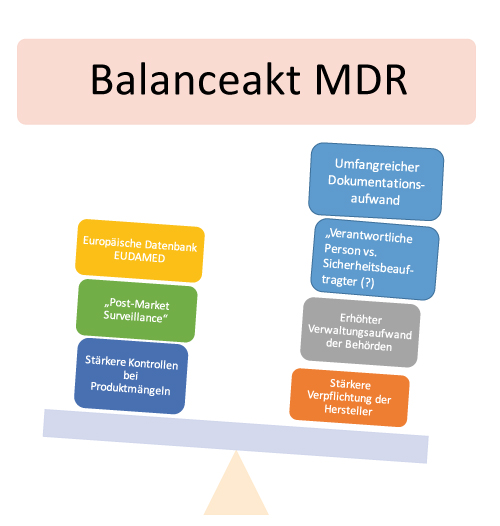 Zukünftig soll eine Expertengruppe in den Zertifizierungsprozess involviert werden, wenn Mängel bei Hochrisiko-Medizinprodukten vorliegen. Außerdem müssen im Rahmen der „Post-Market-Surveillance“ klinische Daten auch nach der Markteinführung gesammelt, dokumentiert und ausgewertet werden. Jedes Medizinprodukt erhält künftig eine Produktidentifizierungsnummer (UDI). Hinzukommt: Eine einheitliche Informationssammlung über umlaufende Medizinprodukte soll die neue Datenbank Eudamed garantieren. Die neue MDR ist eine Herausforderung.
Zukünftig soll eine Expertengruppe in den Zertifizierungsprozess involviert werden, wenn Mängel bei Hochrisiko-Medizinprodukten vorliegen. Außerdem müssen im Rahmen der „Post-Market-Surveillance“ klinische Daten auch nach der Markteinführung gesammelt, dokumentiert und ausgewertet werden. Jedes Medizinprodukt erhält künftig eine Produktidentifizierungsnummer (UDI). Hinzukommt: Eine einheitliche Informationssammlung über umlaufende Medizinprodukte soll die neue Datenbank Eudamed garantieren. Die neue MDR ist eine Herausforderung.
Wirtschaftsakteure stärker in der Pflicht
So haben beispielsweise die Behörden bisher wenig Erfahrung mit der neu geregelten Marktüberwachung. Sie müssen nun stichprobenartig Kontrollen von Produkten und Unterlagen durchführen und die betroffenen Wirtschaftsakteure in die Pflicht nehmen. Die Behörden sind dann dafür zuständig, die Beseitigung der Mängel konsequent zu kontrollieren. Darüber hinaus müssen sie alle beteiligten Stellen informieren und auch Einwendeverfahren verwalten. Bis auf den höheren Verwaltungsaufwand ändert sich aber nicht viel, da das geltende Recht bereits viele Punkte abdeckt. Die Wirtschaftsakteure werden jedoch stärker in die Pflicht genommen, was wiederum neue Ansatzpunkte zur Marktüberwachung durch Behörden ermöglicht.
Rolle der „verantwortlichen Person“ gewinnt an Bedeutung
Kleinere Tücken betreffen beispielsweise die Zukunft des Sicherheitsbeauftragten. Denn die MDR sieht eine „verantwortliche Person“ (responsible person) zur Einhaltung der Regulierungsvorschriften vor. Der Verantwortungsbereich der verantwortlichen Person ist umfassender als der des Sicherheitsbeauftragten. Außerdem ist die Qualifikation spezifischer definiert. Während der Sicherheitsbeauftragte eher über Sachkenntnis verfügen muss, zeichnet die verantwortliche Person eine Fachkenntnis aus. Die Rolle der verantwortlichen Person ist auch deutlich hervorgehobener als die des Sicherheitsbeauftragten, da sie mehr Entscheidungsbefugnisse hat. Je nach Unternehmensgröße dürfen Unternehmen die verantwortliche Person extern benennen. Unklar ist, was das für den bisherigen Sicherheitsbeauftragten bedeutet. Löst die verantwortliche Person den Sicherheitsbeauftragten in Zukunft ab?
Dokumentationsaufwand steigt
Hersteller kritisieren vor allem auch den erhöhten Dokumentationsaufwand von u.a. Entwicklungsakten, Produktakten, Risikomanagementakten. So ist beispielsweise eine umfangreichere klinische Bewertung und ein Nachweis der grundlegenden Anforderungen und Produktbeobachtung notwendig. Die Dokumentation muss immer wieder auf Aktualität geprüft und ergänzt werden. Das frisst Ressourcen.
In den Unternehmen schwirren weiterhin einige Fragezeichen umher. Wer darf eigentlich was? Wer sind die benannten Stellen? Wie wird die MDR von diesen Stellen interpretiert?
Problematisch ist die geringe Anzahl der bisher benannten Stellen. Die Befürchtung ist, dass es nach der Neubenennung der Stellen Anfang 2019 zu einem „Flaschenhals-Effekt“ kommt, da es sowieso seit Jahren eine rückgängige Zahl an benannten Stellen gibt.
Ab dem 26. Mai 2020 ist die MDR verpflichtend anzuwenden. Viele Hersteller kritisieren die nur dreijährige Übergangszeit. Es ist fragwürdig, ob die umfangreichen Anforderungen der MDR in dieser Zeit zu erfüllen sind. Vor allem kleine und mittelständische Unternehmen verfügen oft nicht über die nötigen finanziellen und personellen Ressourcen, um die neuen Anforderungen strukturell im Unternehmen zu verankern.
Gut umgesetzt ist die MDR eine Chance
In den Medien ist immer wieder vereinzelt von Medizinprodukteskandalen zu lesen. Qualitätsmängel an Medizinprodukten sind ein Problem in der Branche und erregen in der Regel viel Aufsehen. Der Grundgedanke der MDR, die Patientensicherheit zu erhöhen und ein europäisches System auf den Weg zu bringen, ist auf jeden Fall positiv. Denn eine Vereinheitlichung und der Versuch, Mängel besser identifizieren zu können, kann unangenehmen Skandalen vorbeugen. Wird die MDR gut umgesetzt, ist sie auf jeden Fall eine Chance, die die Sicherheit der Medizinprodukten fördert. Wie gut diese gute Umsetzung in der knappen Zeit zu bewältigen ist, wird sich erst in naher Zukunft zeigen.